Functional Genomics
Tatsu's paper on modeling TF binding using DNase-seq data is published in Nature Biotechnology
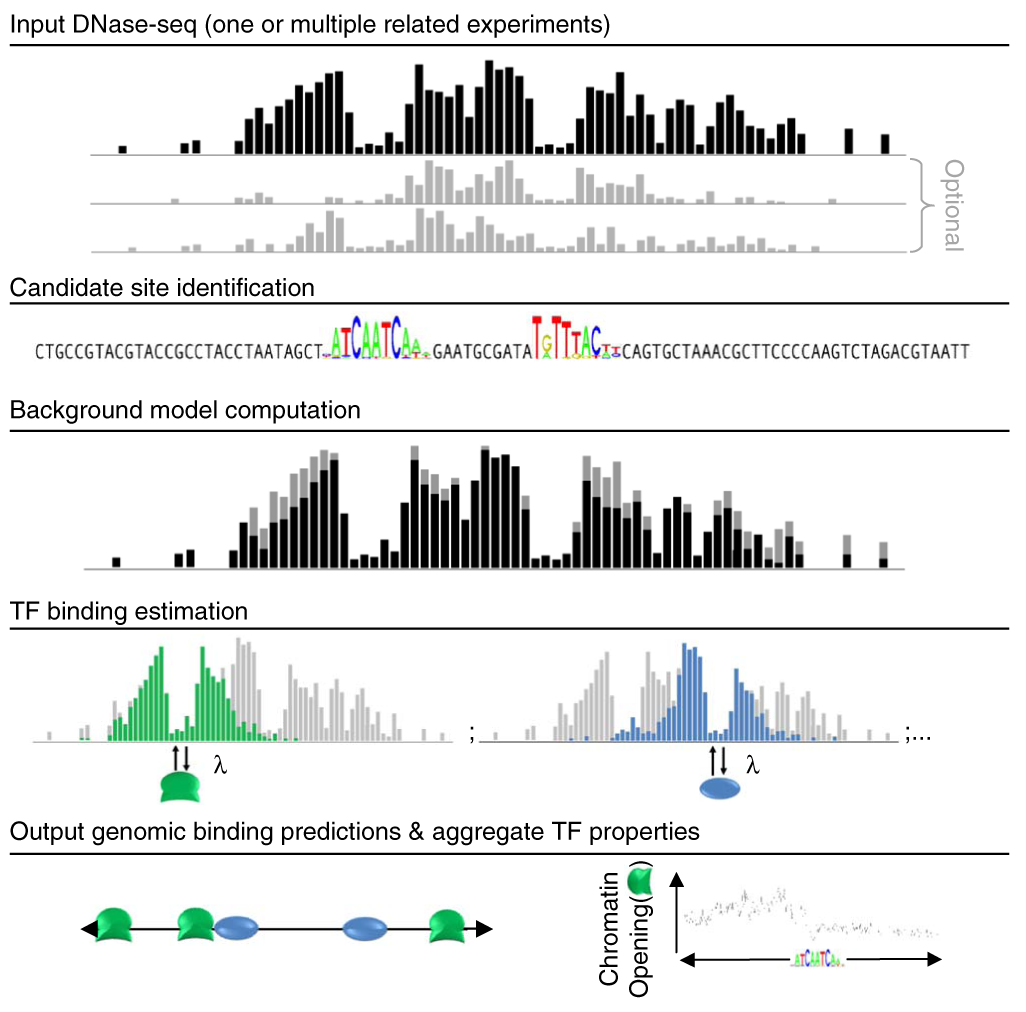
We describe protein interaction quantitation (PIQ), a computational method for modeling the magnitude and shape of genome-wide DNase I hypersensitivity profiles to identify transcription factor (TF) binding sites. Through the use of machine-learning techniques, PIQ identified binding sites for >700 TFs from one DNase I hypersensitivity analysis followed by sequencing (DNase-seq) experiment with accuracy comparable to that of chromatin immunoprecipitation followed by sequencing (ChIP-seq). We applied PIQ to analyze DNase-seq data from mouse embryonic stem cells differentiating into prepancreatic and intestinal endoderm. We identified 120 and experimentally validated eight ‘pioneer’ TF families that dynamically open chromatin. Four pioneer TF families only opened chromatin in one direction from their motifs. Furthermore, we identified ‘settler’ TFs whose genomic binding is principally governed by proximity to open chromatin. Our results support a model of hierarchical TF binding in which directional and nondirectional pioneer activity shapes the chromatin landscape for population by settler TFs.