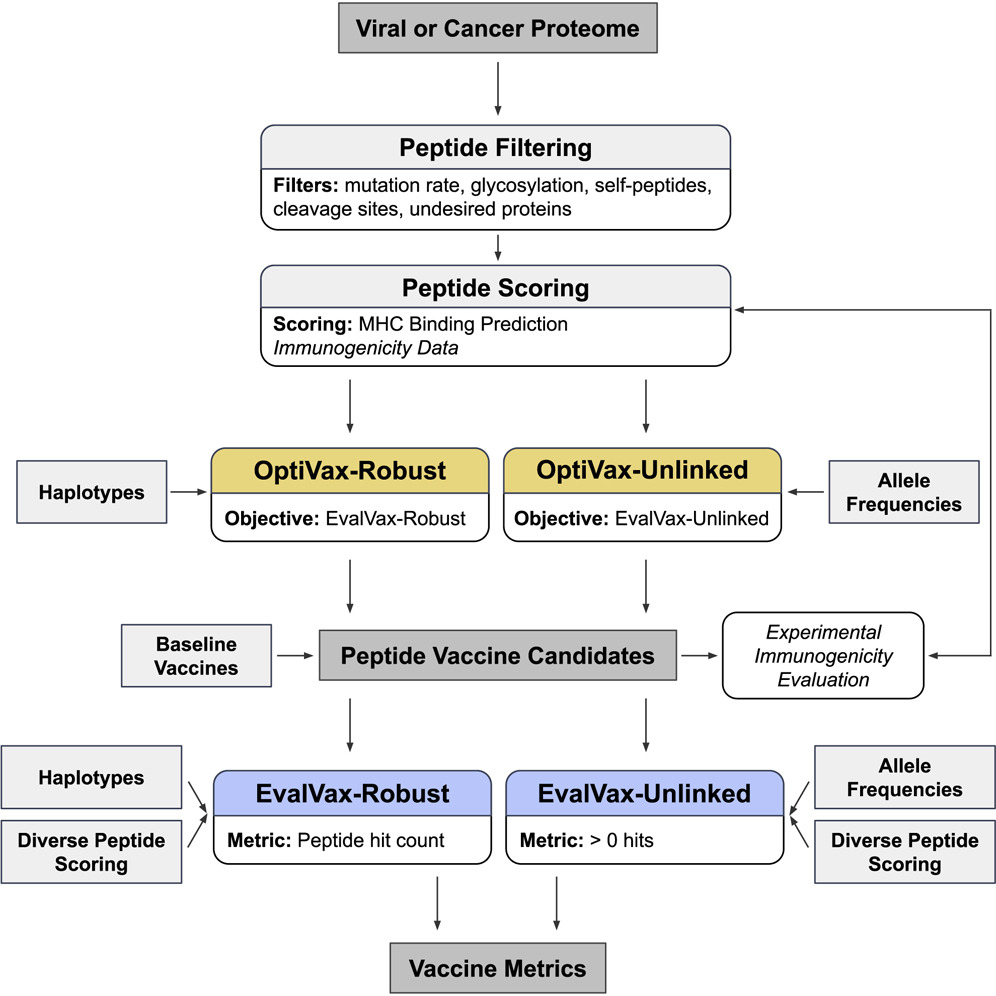
We present a combinatorial machine learning method to evaluate and optimize peptide vaccine formulations for SARS-CoV-2.
We introduce an augmentation strategy for subunit vaccines that adds a small number of SARS-CoV-2 peptides to a vaccine to improve the population coverage of pathogen peptide display.
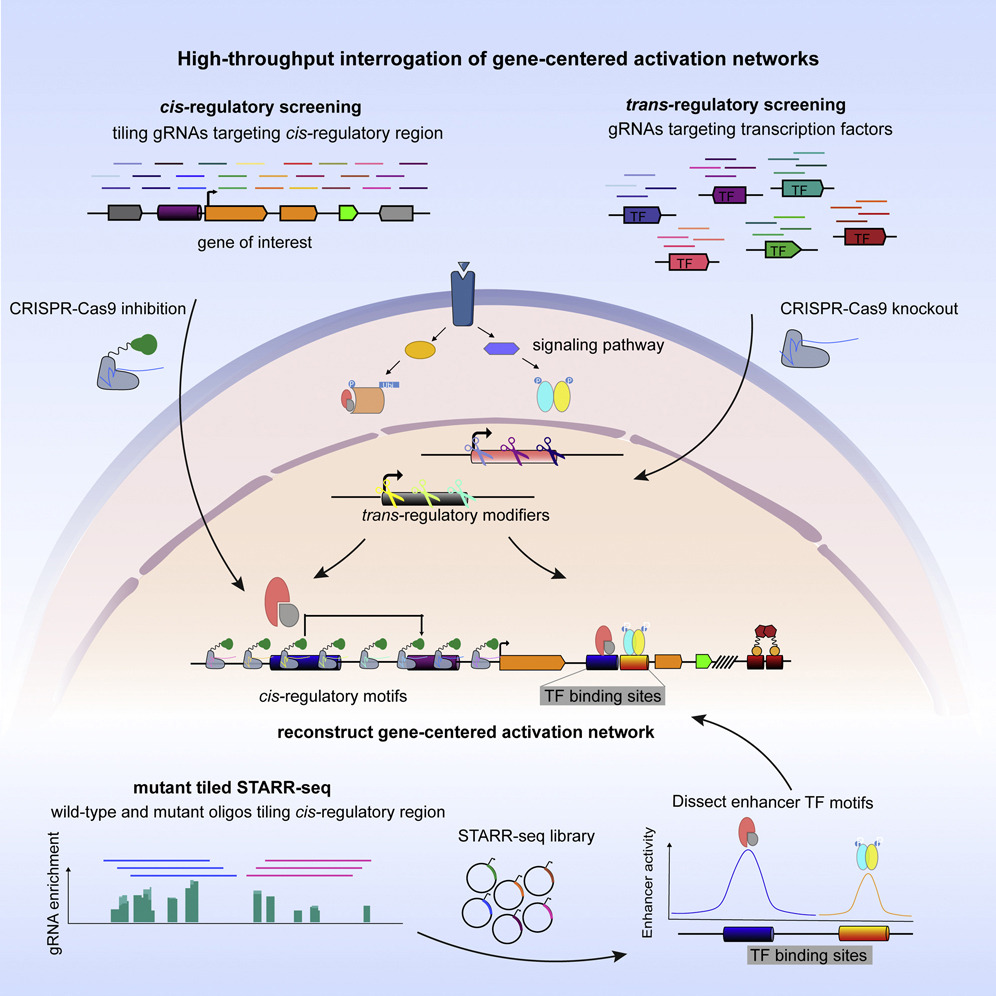
Here we present high-throughput interrogation of gene-centered activation networks (HIGAN), a pipeline that employs a suite of multifaceted genomic approaches to connect upstream signaling inputs, trans-acting TFs, and cis-regulatory elements.